The exchange of anions between the living cell and its environment represents one of the most fundamental life-sustaining phenomena, and many diseases are associated with defected chloride anion transport. Hence, the design of synthetic anion transporters and channels is essential not only for gaining insight on how natural ion carriers and channels work, but also for practical applications such as treatment of channelopathies, supramolecular architecture, anion sensing, catalysis and therapeutics.
A. hetero-Bambusurils
Scheme. 1. hetero-BU[n]s that we prepare by heteroatom displacement approach
B. Studying the unique structure of hetero-Bambusurils
The fused bicyclic structure of glycoluril is a valuable structural motif for a wide variety of cyclic host molecules. The bambusurils (BUs) are comprised of N,N-disubstituted glycoluril units, which are interconnected by only one hoop of single bonds ("single-stranded"). Although being single-stranded, the BUs are relatively rigid molecules due to their subunits high steric demands, which are locked in an alternate arrangement. In the BUs, the glycoluril units present their convex face towards the interior, forming a bamboo-like object. This structure explains why the BUs are anion-binders.
BUs are known to be rigid cavitands that feature an extended, jigger-like conformation, and the BU[6]s strongly bind anions within their hydrophobic cavity. The family of perthio-BUs, does not necessarily share these features. This study reveals that the latter assumes a compact conformation and perthio-BU[6]s are poor anion-binders, crystallizing as anion-free species from solutions containing halide salts. Computational studies show that the equatorial sulfur atoms compete against guest anions for binding with the glycoluril methine groups via strong vdW attractive interactions. These competitive contacts account for the diminished anion-binding of perthio-BUs, account for the diminished anion-binding of perthio-BUs, account for the diminished anion-binding of perthio-BUsaccount for the diminished anion-binding of perthio-BUs and explain their compact conformation.
Scheme 2. Solid-state structures of BU[6] (left) and BU[6] (right) heteroisomers. A. all-oxygen; B. ax-semithio-; C. ax-semiaza-; D. perthio-; E. computed structure of eq-semithio- and F. ax-semiaza-eq-semithio-. All molecules are characterized by a Capped Sticks model whereas all heteroatoms are represented by a Spacefill model (oxygen, red; sulfur, yellow; nitrogen, blue).
C. Unique reactivity and binding properties of sulfur-containing BU[4]s
The semithio- and perthio-BU[4]s form linear coordination polymers with Hg(II) in the solid-state regardless of their intrinsic molecular conformation. The strong involvement of sulfur atoms in intramolecular interactions differentiates the equatorial from the axial (peripheral) heteroatoms, thus offering chemoselective and regioselective transformations.
Fig. 1 Comparison between the crystal structures of the linear chains of Hg(II) complexes with semithio- (left) and perthio-BU[4] (right) in the solid state.
D. From a single anion receptor into a potential anion channel
We show that by converting a semithio-bambus[6]uril into the corresponding semiaza-bambusuril, a single anion receptor is transformed into a potential anion channel. Hence, semiaza-BUs were found to simultaneously accommodate three anions, which were linearly positioned within the cavity along the cavitand's central symmetry axis (Fig. 2).
Fig. 2 Left: Solid-state structure of semithio-BU[6], 43 hosting a bromide anion. Middle and Right: Solid-state structure of two semiaza-bambusurils, 44 (R = H, top) and 44 (R = picolyl; bottom), hosting one bromide (top) or iodide (bottom) and two triflate anions.
The solid state structure of semithio-BU[6] highlighted the bromide-binding site's immediate environment, indicating very close interactions between the anion and the twelve methine hydrogens, which were arranged in two circles above and below the bromide ion. The structures of semiaza-BU[6] are much more interesting in ion transport because the latter can accommodate several anions simultaneously. These complexes demonstrate a remarkable capacity of overcoming the strong electrostatic repulsion among three anions held together at a significantly small distance, as short as 4 Å. It is noteworthy that precisley the same inter-anionic distance of 4 Å was reported for adjacent chloride binding sites in the crystal structures of two mutant E. coli ClC chloride channels.
E. semithio-BU[6]: a transmembrane anion transporter.
Ion transport across the cell membrane is essential for maintaining the cell ion homeostasis and regulating critical processes of life, such as proliferation, differentiation and apoptosis. Generally, nature achieves these tasks using membrane proteins and large macromolecular assemblies. Synthetic small molecules, which function as ion transporters, offer exciting opportunities in biology and medicine. Such molecules could be used as therapeutic agents for treating anion channelopathies. Anion transporters can also induce apoptosis in cancer cells with potential applications in anticancer therapy. In a collaborative study with Prof. Junqiu Liu from Jilin University in China, we demonstrate the transporting ability of semithio-BU[6] as anion carriers through bilayer lipid membrane (POPC) with high selectivity to chlorides over various different anions (Fig. 3).
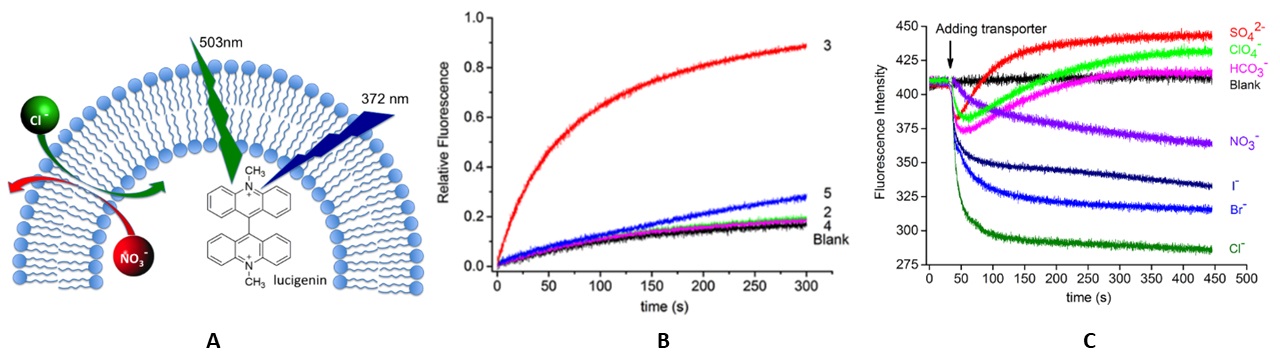
Fig. 3 (A) Schematic view of the Clˉ influx and NO3ˉ efflux across POPC vesicles, monitored by lucigenin fluorescence; (B) chloride transport mediated by various BUs; (C) chloride transport of semithio-BU[6] (10 ml in DMSO solution, 2.7 mol%) using the potential-sensitive dye safranin O (Ex. 480 nm, Em. 520 nm) with various anions (DMSO was used as blank).
semithio-BU[6] is a significantly more efficient chloride transporter through bilayer lipid membranes than all other analogs due to their significant lipophilic variability. The transport efficiency was independent of the cation identity, indicating that the observed phenomena reflect anion-anion antiport rather than a symport mechanism. Moreover, semithio-BU[6] operates as a uniport transporter of chloride anions in the presence of phosphate and bicarbonate anions. These remarkable findings reflect this carrier's ability to act as a valinomycin-like transporter, which could be exploited in biomedical applications..
F. Anchoring and packing of self-assembled monolayers (SAMs) of semithio-BUs on gold surface
Much attention has been received to thiolated SAMs because of their electrochemical sensing behavior and molecular electronic properties. Opposed to thiols, thiocarbonyl binding to gold surface results in forming a new bond rather than the replacement of an existing one (i.e., from RS-H to RS-Au). Surprisingly, SAMs based on macrocycles with docking thiocarbonyl groups have rarely studied. However, small closed-shell molecules with thiocarbonyl docking groups have shown induced radical character upon binding to the gold surface leading to higher conductance than thiolate analog.
In a collaborative study with Prof. Jurriaan Huskens from the University of Twente, Netherlands, a range of physicochemical characterization techniques combined with STM and MD simulations showed that semithio-BU[4] and semithio-BU[6] undergo significant conformational rearrangements upon attachment to a gold surface (Fig. 4). The molecules attach using most of the S atoms, with the energetic benefit of Au-S binding compensating for any steric strain. Accordingly, the molecule is forced to adopt a more confined/pinned conformation on the surface.
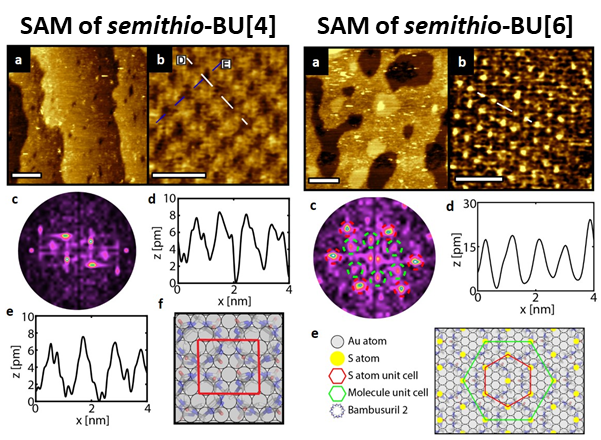
Fig. 4 STM images of a SAM of semithio-BU[4] (Left) and semithio-BU[6] (Right) on Au(111). Left: (a) Large scale topographic image (100 x 100 nm2, scale bar is 20 nm), taken at 250 pA and 700 mV. (b) STM image of a rectangular ordered domain (5.2 x 5.2 nm2, scale bar 2 nm). (c) The corresponding FFT of (b) showing rectangular symmetry. (d-e) STM height profile of the white and blue line in (b). (f) Proposed 4x2√3 unit cell (red rectangle, 1.15 x 1 nm2) of semithio-BU[4] on the unreconstructed Au(111) surface. Right: (a) Large scale topographic image (100 x 100 nm2, scale bar is 20 nm), 190 pA and 600 mV. (b) STM image of a hexagonal ordered domain (9.2 x 9.2 nm2, scale bar 3 nm). (c) The corresponding FFT of (b) showing the same hexagonal symmetry with a 0.86 nm periodicity (red) and hexagonal symmetry with a periodicity of approximately 1.5 nm (green). (d) STM height profile of the white line in (b) confirming a 0.86 nm distance between the sulfur atoms. (e) Proposed (3√3x3√3)R30o unit cell (green hexagon) of semithio-BU[6] on Au(111) with the (3x3) sulfur hexagon marked in red (0.86 x 0.86 nm2).
G. Inherently chiral bambusurils
Chirality in macrocycles with large cavities has attracted significant attention due to their extensive use in chiral recognition, especially in their role in resolving racemates and in selectively transporting chiral guests through bulk liquid membrane technologies. Bambusurils (BUs) are rigid achiral macrocycles, exhibiting Sn point group of symmetry. A general criterion for chirality is the absence of any improper symmetry elements, i.e. Sn, including mirror image s, or a center of inversion, i. As such, these cavitands can be made chiral by either appending an intrinsically chiral functionality to the macrocyclic backbone or by using a non-symmetric subunit to produce stereogenic cavitands (Scheme 2). Therefore, designing inherently chiral hetero-BUs would be appealing for developing applications in the field of chiral sensing technologies. Recently, we introduced a new class of bambus[4]urils (BU[4]s) composed of asymmetric N,N’-disubstituted glycoluril subunits with different alkyl groups were prepared. Accordingly, four macrocyclic diastereoisomers are possible: two Sn symmetric achiral macrocycles and two macrocycles that are “inherently” chiral (Scheme 3). Indeed, if one of the methyl groups in glycoluril is substituted by a different group, then a mixture of two enantiomers is obtained. The cyclotetramerization of the glycoluril racemate with paraformaldehyde generates four possible stereoisomers of R'4Me4BU[4] (R'¹Me).
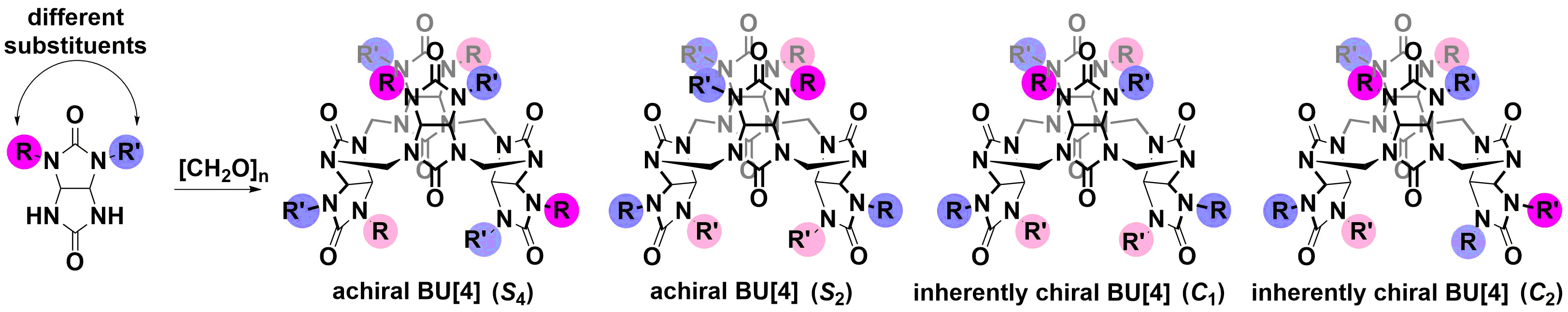
Scheme 3. BU[4] exhibiting four possible diastereoisomers: two of which are symmetric achiral (S4 and S2 symmetry), and two "inherently" chiral macrocycles (C1 and C2 symmetry).
The relative "head-to-tail" arrangement of the N-substituents in Bn4Me4BU[4] with S4 symmetry was fully confirmed by X-ray spectroscopy analysis. Chiral HPLC resolved the chiral Pr4Me4BU[4] (C1 symmetry into its enantiomers. All four inherently chiral bambusuril pairs of Pr4Me4BU[4] and Bn4Me4BU[4] stereoisomers were resolved by 1H NMR spectroscopy with the aid of (R)-BINOL as a chiral solvating agent. This latter methodology provides a rapid and robust approach for investigating inherently chiral cavitands' enantiopurity, which complements conventional chromatographic techniques.
H. Cucurbiturils
We have also focused on the binding properties of cucurbit[6]uril (CB[6]). This cavitand has attracted our attention for several years because it features a highly symmetric structure with high electron density at the carbonyl oxygen atoms on the cavitand rims and clearly illustrates its cation-receptor functionality. The cavitand's entry also leads to an entirley hydrophobic interior, which resulted in threading of neutral, highly hydrophobic alkyl chains. In this context, we prepared five homologous bis-α,ω-azidoethylammonium alkanes, where the number of methylene groups between the ammonium groups ranges from 4 to 8 (Fig. 5 top: complexes of 2-6@CB[6]). While the distance between the portal plane and most atoms at the guest end-groups increases progressively with the molecular size, the β-nitrogen atoms of the azide groups maintain a constant distance from the portal plane in all homologues, pointing at a strong attractive interaction between the azide group and the portal (Fig. 5 bottom).
In collaboration with the research group of Prof. Michael K. Gilson (UCSD), we were able to show by quantum computational analyses that strong electrostatic interactions in the form of orthogonal dipole−dipole interaction are the main driver for this attraction. In contrast, the alternative mechanism of n → π* orbital delocalization did not seem to play a significant role in this interaction. The computational studies also indicate that the interaction is not limited to azides, but generalizes to other isoelectronic heteroallene functions, such as isocyanate and isothiocyanate.
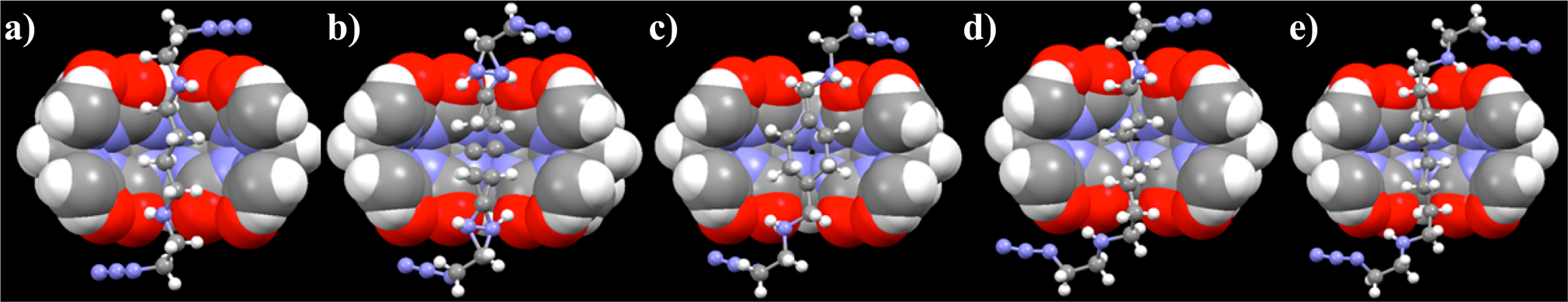
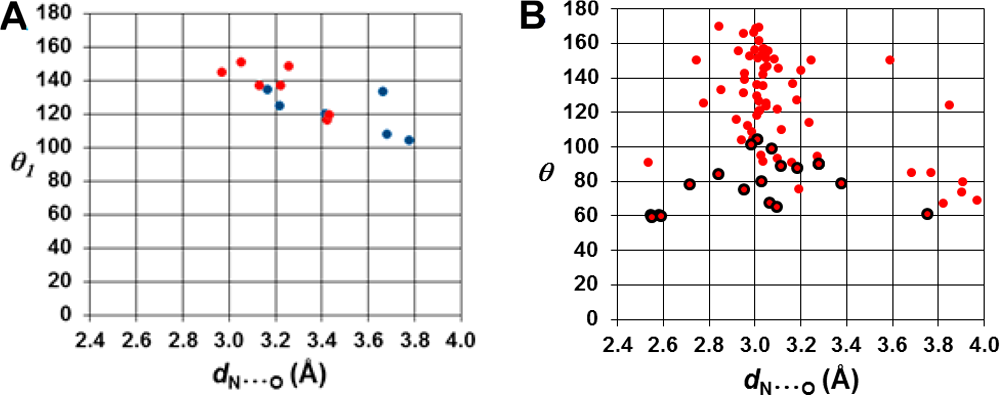
Fig. 5. Top: X-ray crystal structures of (a) 2@CB[6], (b) 3@CB[6], (c) 4@CB[6], (d) 5@CB[6], and (e) 6@ CB[6]. CB[6] is presented in a cross-sectional, space-filling format. Atom doubling and missing bonds indicate disordered structures; Left bottom: Scatter plot correlation between θ (deg) and dN···O (Å), extracted from the X-ray structural data. The red circles refer to the interactions with the azide β-nitrogen atom, and blue circles refer to the γ-nitrogen; Right bottom: Scatter plot correlation between θ and dN···O (all referring to the azide β-nitrogen) extracted from the CSD database. The red circles refer to intramolecular interactions, whereas the red circles with a dark margin describe intermolecular interactions.
I. Multifarenes
The most common cavitands, are all homo-cyclooligomers of a single building block. Consequently, their binding properties, substitution patterns, functionalization, and solubility reflect the monomeric unit's characteristics. In contrast, the hetero-cyclooligomeric cavitands multifarenes (MFs), which comprise multiple building blocks, are much more versatile in shape, size, rigidity and solubility and binding properties (Scheme 4).
Scheme 4. Top: General design elements of MF[2,2] and Bottom: Gradual evolution of curved multifarenes by stepwise synthetic modifications
Recently, we reported the stepwise evolution of curved multifarene structures from planar precursors and increasing the extent of curvature in a controlled manner. These synthetic steps highlight three architectural design elements: a) employment of different aromatic units and changing their relative orientation, b) changing the hybridization of the linking atoms from sp2 to sp3, and c) rigidification of the non-planar system by 5-membered rings. These design elements are reminiscent of those used to transform flat graphene sheets into curved carbon structures, such as fullerenes and carbon nanotubes.